分析技术研习室
课题组每周研讨会
第八期 WES 变异检测-GATK流程
Dnaseq and mutation calling
总流程
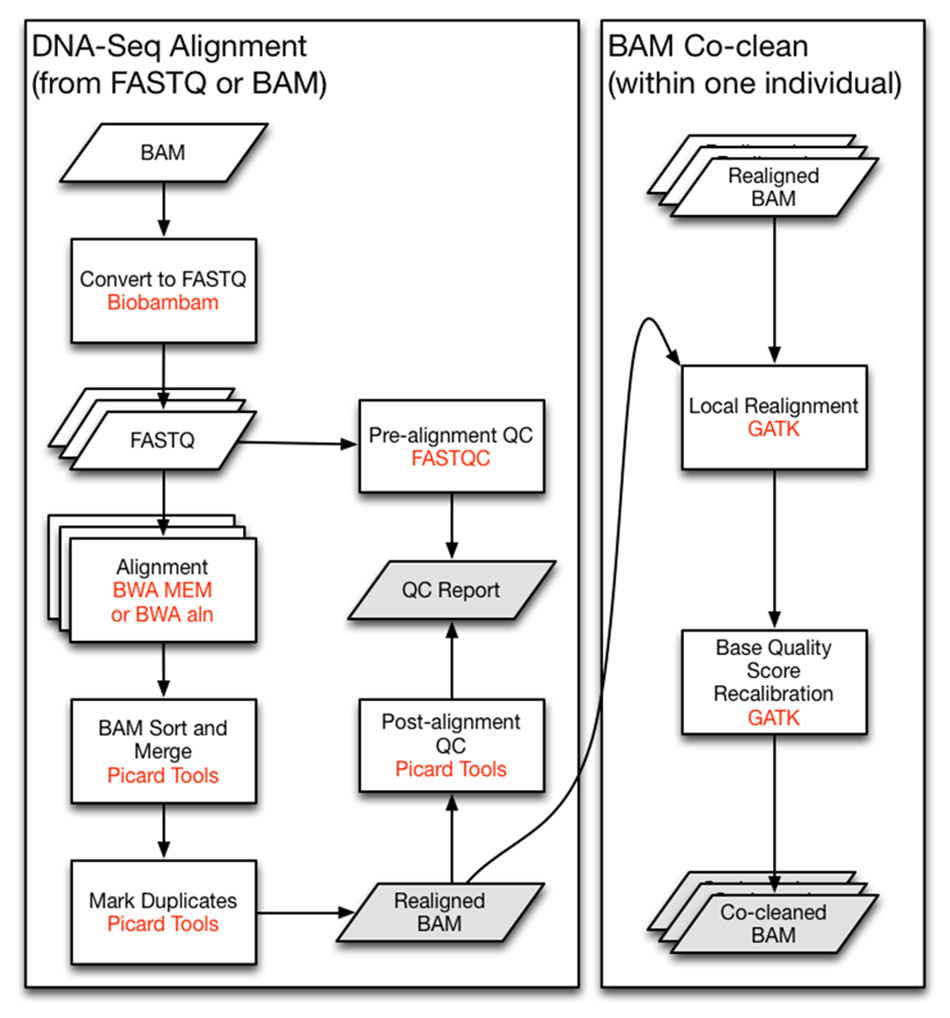

1.TCGA 原始测序数据下载(师兄原来下载好的)
2.原始数据解压 sra解压为fastq
所用的工具是sra-toolkit里的fastq-dump,下载用conda下载
fastq-dump -outdir fastq --split-3 --skip-technical --clip --gzip $workdir/sra/<sample>.sra
–outdir
–gzip 输出结果使用gzip压缩
–skip-technical 只输出biological reads,不然会把technical reads输出
technical reads: (adapters, primers, barcodes)
–split-files 把pair-end测序分成两个文件输出,正向反向的测序数据分开
–split-spot 将reads分为左端右端测序数据,输出到一个文件中
–split-3 将reads分为左端右端测序数据,如果只有单端序列的话 ,将会产生一个文件
–clip 去除标签
3.FASTQC 测序质量检验
fastqc -o ~/dnaseq/fqc/ -t 8 ~/dnaseq/fastq/<sample>
4.multiqc
整合所有样本的质控数据
关注点
-
Per Sequence quality scores序列测序质量
查看碱基质量是否存在普遍过低的情况,横坐标是序列平均碱基质量值,纵坐标为序列的数量。碱基序列平均质量值大于30,可判断质量较好。
Q=-10*log10(error P) ,Q20为1%的错误率
-
Per Base Sequence Content 碱基测序含量 统计序列的每个位置四种不同的碱基占总碱基数的比例,正常情况下碱基出现的频率应该是差不多的。
-
Per Sequence GC Content GC含量
GC含量分布是正态分布,如果出现两个或者多个峰值,可能数据中有其他来源的序列污染。
-
Per Base N Content 每个碱基N的位置
测序仪器不能识别某个位置是哪个碱基时,用N表示,正常情况下N的比例很小。
5.去接头
Trim galore适用于所有高通量测序,包括RRBS(Reduced Representation Bisulfite-Seq ), Illumina、Nextera 和smallRNA测序平台的双端和单端数据。主要功能包括两步: 第一步首先去除低质量碱基,然后去除3’ 末端的adapter, 如果没有指定具体的adapter,程序会自动检测前1million的序列,然后对比前12-13bp的序列是否符合以下类型的adapter:
Illumina: AGATCGGAAGAGC
Small RNA: TGGAATTCTCGG
Nextera: CTGTCTCTTATA
下载安装trim galore
conda安装
使用
trim_galore -q 20 --phred33 --stringency 3 --length 20 -e 0.1 --paired --gzip $workdir/<sample>_1.fastq.gz $workdir/<sample>_2.fastq.gz -o fqclean
–quality:设定Phred quality score阈值,默认为20。 –phred33:表示测序平台使用的Phred quality score。 –stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1。 –length:设定输出reads长度阈值,小于设定值会被舍弃 –paired:对于双端测序结果,一对reads中,如果有一个被舍弃,那么另一个也会舍弃。 –gzip:数据zip打包 –output_dir:输出目录
error
cutadapt: error: Error in FASTQ file at line 385083733: Premature end of file encountered. The incomplete final record was: '@SRR31'
6. bwa 比对
这一步的意义是将测序数据与参考基因组mapping,参考基因组是hg38。 用到的工具是:BWA,安装依旧使用的conda BWA流程: BWA的比对有三个不同的算法: BWA-backtrack:是用来比对ILLUMINA的序列,reads长度能达到100bp BWA-SW:用于比对long-reads的,支持的长度为70bp-1Mbp;同时支持剪切性比对 BWA-MEM:是更新的算法,也是这次我们要用的算法,支持较长的reads同时也支持剪接性比对。
6.1使用:
进行比对之前,需要对fastq文件构建索引。算法选用的bwtsw
bwa index [ –a ]
OPTIONS:
| -a [is | bwtsw] 构建index的算法,有两个算法: is 是默认的算法,虽然相对较快,但是需要较大的内存,当构建的数据库大于2GB的时候就不能正常工作了。 bwtsw 对于短的参考序列式不工作的,必须要大于等于10MB, 但能用于较大的基因组数据,比如人的全基因组。 |
6.2 mem比对
bwa mem [options] ref.fa reads.fq [mates.fq]
bwa mem -M -R "@RG\tID:<sample>\t\
LM:<sample>\t\
SM:<sample>\t\
PL:illumina\tPU:<sample>"\
/public/home/GRCh38.d1.vd1.fa\
/public/home/<sample>_1_val_1.fq.gz\
/public/home/<sample>_2_val_2.fq.gz\
><sample>.sam
具体参数含义:
-M 将 shorter split hits 标记为次优,以兼容 Picard’s markDuplicates 软件
-R STR 完整的read group的头部,可以用 ‘\t’ 作为分隔符, 在输出的SAM文件中被解释为制表符TAB. read group 的ID,会被添加到输出文件的每一个read的头部。
-R STR Complete read group header line. ’\t’ can be used in STR and will be converted to a TAB in the output SAM. The read group ID will be attached to every read in the output. An example is ’@RG\tID:foo\tSM:bar’.
比对这一步有一个非常重要的点就是参考基因组的染色体排序问题,我的参考基因组排序是乱的导致后面运行的时候报错
基于提供的ref.fa文件中的chromosome/contig的顺序。当你使用比对工具将fastq文件中的reads比对上参考基因组后会生成SAM文件,SAM文件包含头信息,其中有以@SQ开头的头信息记录,reference中有多少条chromosome/contig就会有多少条这样的记录,而且它们的顺序与ref.fa是一致的 sam bam 进阶
7.redor 排序
7.1
排序之前需要用samtools构建参考序列的索引
samtools faidx hg38.fa
生成hg38.fa.fai文件
7.2 选做
由BWA生成的sam文件时按字典式排序法进行的排序(lexicographically)进行排序的(chr10,chr11…chr19,chr1,chr20…chr22,chr2,chr3…chrM,chrX,chrY),但是GATK在进行callsnp的时候是按照染色体组型(karyotypic)进行的(chrM,chr1,chr2…chr22,chrX,chrY),因此要对原始sam文件进行reorder。可以使用picard-tools中的ReorderSam完成。
java -jar picard.jar ReorderSam\
I=<sample>.sam\
O=<sample>.resort.sam\
REFERENCE=hg38.fa
8.sam转bam
samtools view -bS <sample>.sam> <sample>.bam
9.bam 排序
java -jar -Xmx12G -Djava.io.tmpdir=tmp picard.jar SortSam\
I=<sample>.bam\
O=<sample>.sort.bam\
SORT_ORDER=coordinate
检查排序
10.Mark Duplicates
标记PCR重复,在之前的质量控制中会看到有很多重复序列,也就是在测序是经过PCR产生的重复,需要将其标记出来(不用去除)后续GATK会识别这些标记。 详细解释
工具:picard中的MarkDuplicates
java -jar picard.jar MarkDuplicates\
I=<sample>.sort.bam\
O=<sample>.rdup.bam\
VALIDATION_STRINGENCY=LENIENT \
MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000 \
M=<sample>.sort.addhead.rmdup.metric
11.标记之后生成索引.bai文件。
samtools index <sample>.rdup.bam <sample>.rdup.bam.bai
12.BQSR(Base Quality Score Recalibration)
工具:GATK 这一步是对bam文件里reads的碱基质量值进行重新校正,使最后输出的bam文件中reads中碱基的质量值能够更加接近真实的与参考基因组之间错配的概率。这一步适用于多种数据类型,包括illunima、solid、454、CG等数据格式。在GATK2.0以上版本中还可以对indel的质量值进行校正,这一步对indel calling非常有帮助。 具体信息GATK 分为两步: BaseRecalibrator Applybam
sample=<sample1>
dbsnp=/ftp.broadinstitute.org/bundle/hg38/dbsnp_146.hg38.vcf.gz
dbsnp1000G=/ftp.broadinstitute.org/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz
dbindel100G=/ftp.broadinstitute.org/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
if [ -e $sample.rdup.bam ]; then
java -jar -Xmx12G -Djava.io.tmpdir= tmp \
gatk-package-4.1.3.0-local.jar BaseRecalibrator \
-R GRCh38.d1.vd1.fa \
-I ${sample}.rdup.bam \
-O ${sample}.recal_data.table \
--known-sites $dbsnp --known-sites $dbsnp1000G --known-sites $dbindel100G && \
java -jar -Xmx12G -Djava.io.tmpdir=tmp gatk4-4.1.3.0-0/gatk-package-4.1.3.0-local.jar ApplyBQSR \
-R GRCh38.d1.vd1.fa \
-I ${sample}.rdup.bam \
--bqsr-recal-file ${sample}.recal_data.table \
-O ${sample}.sorted.marked.BQSR.bam
echo gatk-BQSR `date`
fi
Mutation calling
Somatic mutations were detected by following GATK best practice with Mutect2 [@van2013fastq].
1. call somatic SNVs and short INDELs
gatk --java-options "-Xmx20G -Djava.io.tmpdir=/public/home/liuxs/lhm/tmp" Mutect2 \
-R GRCh38.d1.vd1.fa \
-L Homo_sapiens_assembly38.targets.interval_list \
-I <sample1>.sorted.marked.BQSR.bam \
-tumor <sample1> \
-I <sample2>.sorted.marked.BQSR.bam \
-normal <sample2>\
--germline-resource af-only-gnomad.hg38.vcf.gz \
--panel-of-normals 1000g_pon.hg38.vcf.gz \
--af-of-alleles-not-in-resource 0.0000025 \
--disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \
--bam-output <sample>.bam \
--f1r2-tar-gz <sample>.tar.gz \
-O <sample>.vcf
2. Estimate cross-sample contamination using GetPileupSummaries and CalculateContamination.
gatk --java-options "-Xmx20G -Djava.io.tmpdir=tmp" GetPileupSummaries \
-I <sample2>.sorted.marked.BQSR.bam \
-L Homo_sapiens_assembly38.targets.interval_list \
-V af-only-gnomad.hg38.vcf.gz \
-O <sample2>-normal.pileups.table
gatk --java-options "-Xmx20G -Djava.io.tmpdir=tmp" CalculateContamination \
-I <sample1>-tumor.pileups.table \
-matched <sample2>-normal.pileups.table \
-O <sample>-contamination.table \
-tumor-segmentation <sample>-segments.table
3. Get tumor artifacts using LearnReadOrientationModel.
java -jar gatk-package-4.1.3.0-local.jar LearnReadOrientationModel \
-I <sample>.tar.gz \
-O <sample>-tumor-artifact-prior.tar.gz
4. Keep confident somatic calls using FilterMutectCalls.
gatk --java-options "-Xmx20G -Djava.io.tmpdir=tmp" FilterMutectCalls \
-R GRCh38.d1.vd1.fa \
-V <sample>.vcf \
-O <sample>.filter.vcf \
--tumor-segmentation <sample>-segments.table \
--contamination-table <sample>-contamination.table \
--ob-priors <sample>-tumor-artifact-prior.tar.gz \
--min-allele-fraction 0.05
5. get PASS labeled mutation
可以自己筛选也可以用工具vcftools
vcftools --vcf <sample>.filter.vcf --remove-filtered-all --out <sample>-SNvs_only
6. annotate mutations
This step annotated somatic mutations with VEP [@mclaren2016ensembl].
vep --input_file <sample>-SNvs_only.recode.vcf \
--output_file <sample>.anno.vcf \
--symbol --term SO --format vcf --vcf --cache --assembly GRCh38 --tsl \
--hgvs --fasta ~/.vep/homo_sapiens/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz \
--offline --cache --dir_cache ~/.vep/ \
--transcript_version --cache_version 98 \
--plugin Downstream --plugin Wildtype \
--dir_plugins ~/.vep/Plugins