分析技术研习室
课题组每周研讨会
eQTL计算原理
全基因组关联分析(GWAS)
定义:对于多个个体在全基因组范围的遗传变异多态性(SNP)进行检测,获得基因型,将基因型与表型进行统计学分析,根据显著性等关系筛选出最有可能影响该性状的遗传变异,目的是通过这种方法找出与变异相关的基因。
Expression Quantitative Trait Loci(eQTL) analysis
表达数量位置的基因座,它指的是染色体上一些能特定调控mRNA和蛋白表达水平的区域,其mRNA/蛋白质的表达水平量与数量性状成比例关系。eQTL analysis是将基因表达水平的变化和基因型连接起来,研究遗传突变与基因表达的相关性。
eQTL可以分为顺式和反式两种
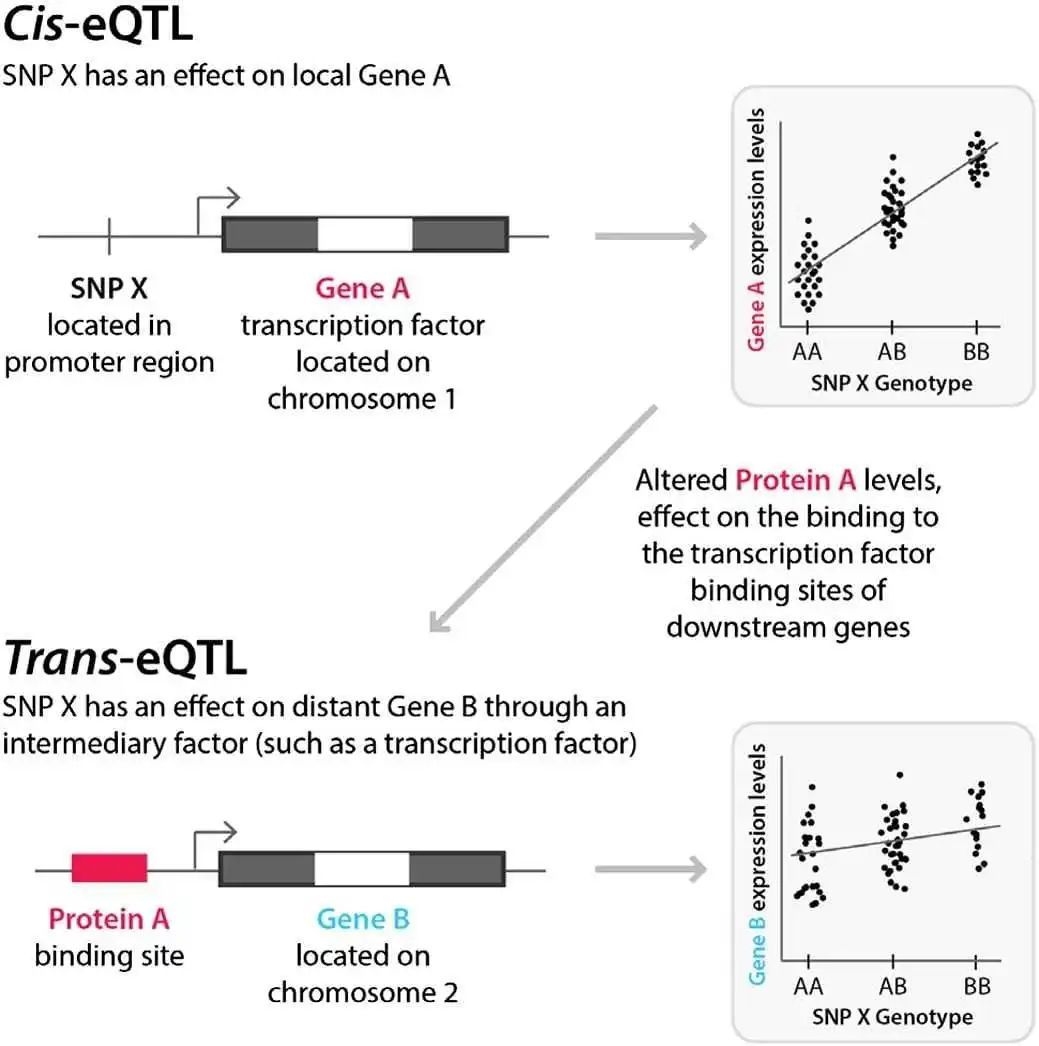
eQTL分析的本质是以全部的DNA变异位点为自变量,轮流以每种mRNA表达量为因变量,用大量的个体数据做样本进行线性回归,得到每一个SNP位点和每一个mRNA表达量间的关系。
线性回归(linear regression)
回归指的是在数据建模中因变量是定量变量。
线性回归是指,对于自变量 $x$和因变量$y$可以写成下列的线性模型: \(Y= \mu + \beta X +\epsilon\) 其中$\mu$是截距,$\beta$是斜率,$\epsilon$是随机误差。
我们要求的是,对于给定的数据点($x_1,y_1$),($x_2,y_2$),…,($x_n,y_n$),存在一个合适的直线\(\hat{y}= \hat{\mu} +\hat{\beta}x\)能更好的拟合我们的数据,最长使用的是最小二乘回归:每个样本点到这条直线的数值距离的平方和最小。最小二乘回归也就是求残差平方和(SSE)的最小值。
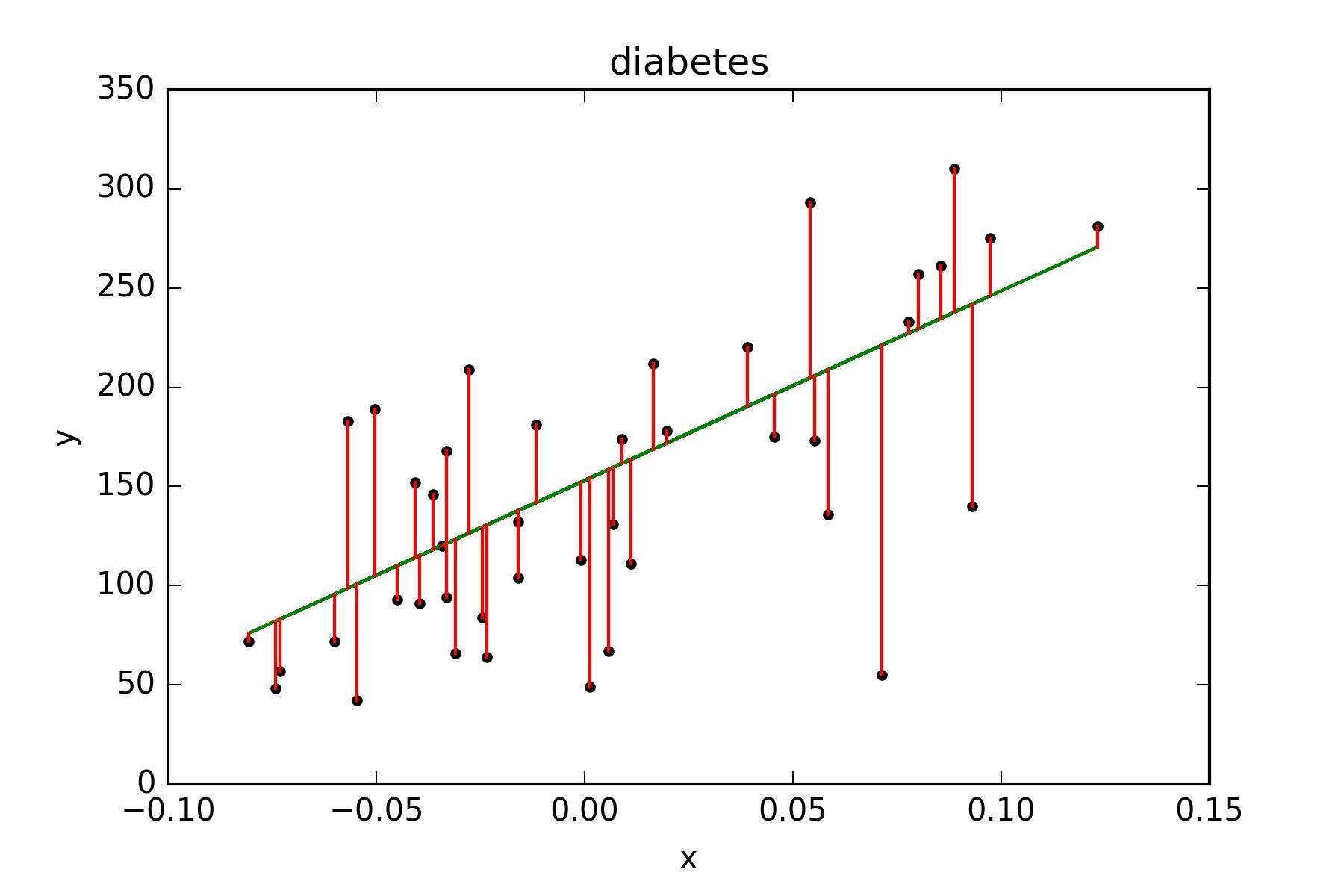
其他的回归方式:多元线性回归,方差分析等。
实际应用
在做eQTL分析时,对于回归模型$Y= \mu + \beta X +\epsilon$其中:
$Y$指的是基因的表达水平;
$X$指的是考虑的SNP的基因类型,编码为0,1,2;
$\mu$指的是AA基因型的表达水平;
$\beta$指的是每一个等位基因B对Y的表达有多大的影响;
$\epsilon$指的就是随机误差。
Matrix eQTL
以该方法为例,这是一个研究团队在12年提出的一个R包,相较于其他的的计算方法,它在运行时间上是更有优势的。
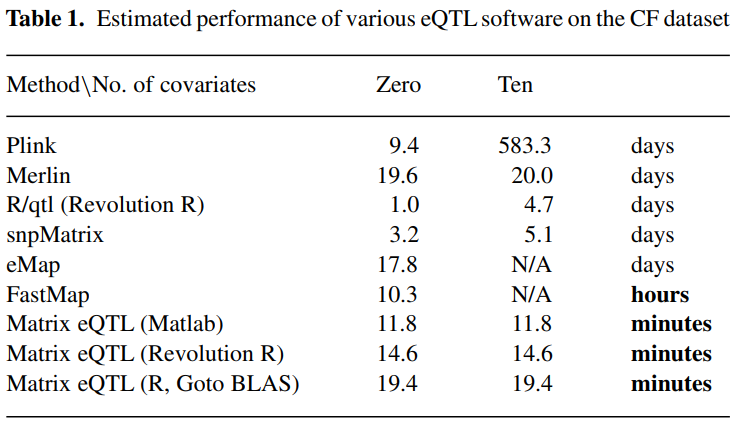
这个R包需要genotype,expression,gene location, SNP location,covariates.协变量covariates指的是血型,年龄,性别这些因素。
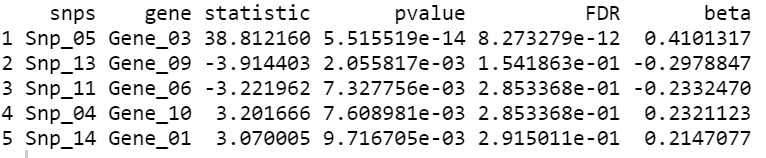
用作者给提供的数据演示,可以得到以下的结果
与癌症相关的研究
因为是想将eQTL的计算方法应用到癌症研究的领域,所以在查找文献时我找到了两篇文献使用这种方法;
第一个是作者使用METABRIC,TCGA,GTEx数据库里面的乳腺癌位点相关的数据做的全基因组关联分析,他做的是顺式的eQTL分析,作者使用的是线性模型,但是没给出一个具体的方式。
而另一篇文献是比较值得借鉴的,作者是用eQTL分析获得乳腺癌的危险位点。在eQTL分析是: \(T_i = Sc_i+M_i+\epsilon_i\\\epsilon_i=G_i+\omega_i\) $T$是转录丰度;$G$是种系基因型;$Sc$是体细胞拷贝数影响;$M$是启动子区域的CpG甲基化水平。作者的分析策略是:通过多元线性回归去计算$Sc_i$和$M_i$的残差表达式$\epsilon_i$,然后再回归残差表达式到种系基因型$G_i$。
作者通过这种方式来分别估计了遗传决定因素,体细胞copynumber变化,和甲基化水平对转录丰度的影响。