分析技术研习室
课题组每周研讨会
Hic数据介绍及相关分析
1,什么是Hic数据?
Hi-C是研究染色质三维结构的一种方法。Hi-C技术源于染色体构象捕获(Chromosome Conformation Capture, 3C)技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。
|  |
2,Hic数据的优势
- 通过Scaffold间的交互频率大小,可以对已组装的基因组序列进行纠错。
- 基因信息不再仅仅是contig片段,而是被划分至染色体上,成为染色体水平。
- 无需辛苦的构建群体,单一一个体就能实现染色体定位。
- 相比遗传图谱,标记密度更大,序列定位更完整。
- 可以开展染色体重排等结构变异研究。
- QTL、GWAS可以定位区间到某个染色体。
- 可以解析该物种的三维基因结构、染色体互作及动态变化。
3,目前的处理流程
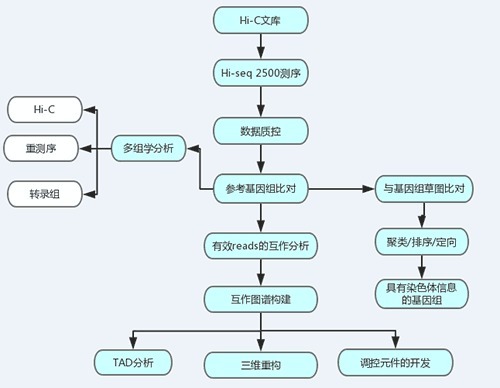
4,分析主要工具
目前针对Hi-c数据处理的工具主要是Hic-pro和juicer
|  |
5,juicer的安装及使用
juicer由两部分组成:从原始数据到创建Hi-C文件的pipeline和后续分析工具。
-
首先要安装依赖软件,
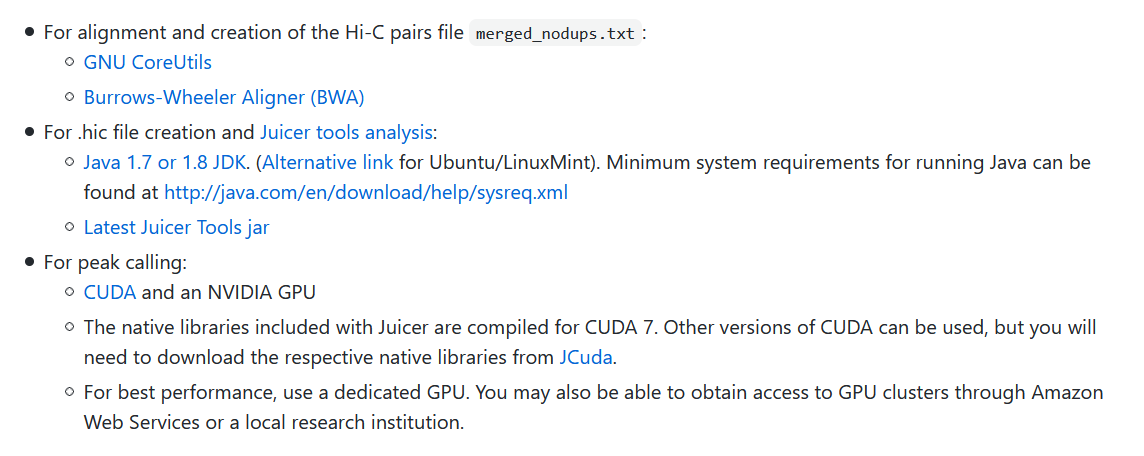
- 注意“bwa”一定要是最新的版本。
PATH=/public/home/wangshx/nw/package/bwa-0.7.17/:$PATH
-
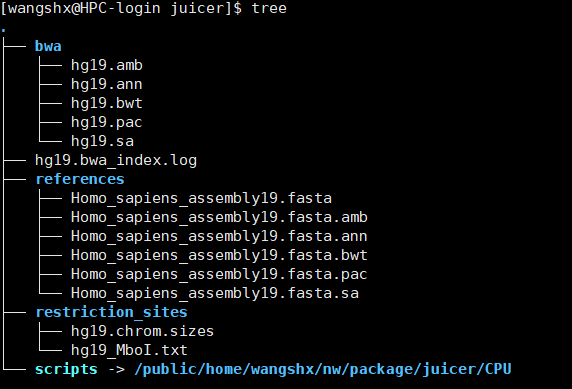
新建juicer目录,构建参考基因组及索引所在目录,并下载相关数据。
mkdir /home/juicer
cd juicer
mkdir references; cd references
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta.amb
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta.ann
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta.bwt
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta.pac
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/references/Homo_sapiens_assembly19.fasta.sa
cd ..
-
构建参考基因组酶切图谱所在目录,并下载相关数据。
mkdir restriction_sites; cd restriction_sites
wget https://s3.amazonaws.com/juicerawsmirror/opt/juicer/restriction_sites/hg19_MboI.txt
awk 'BEGIN{OFS="\t"}{print $1, $NF}' hg19_MboI.txt > hg19.chrom.sizes
cd ..
-
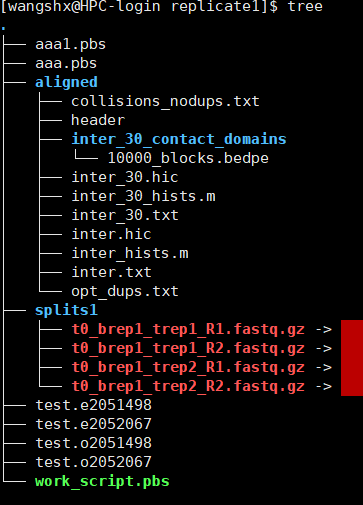
构建原始测序数据所在目录,并下载相关数据(测试数据)可选择。文件夹名字必须是fastq。
mkdir HIC003; cd HIC003
mkdir fastq; cd fastq
wget http://juicerawsmirror.s3.amazonaws.com/opt/juicer/work/HIC003/fastq/HIC003_S2_L001_R1_001.fastq.gz
wget http://juicerawsmirror.s3.amazonaws.com/opt/juicer/work/HIC003/fastq/HIC003_S2_L001_R2_001.fastq.gz
cd ..
测序数据的名称一定要匹配,如:
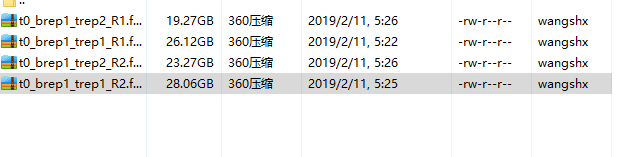
-
新建目录下载juicer(十分重要)
mkdir /home/package
cd package
git clone https://github.com/theaidenlab/juicer.git
-
回到原来的目录,并建立juicer的软连接
cd /home/juicer
ln -s /home/juicer/CPU scripts
cd scripts/common
wget https://hicfiles.tc4ga.com/public/juicer/juicer_tools.1.9.9_jcuda.0.8.jar
ln -s juicer_tools.1.9.9_jcuda.0.8.jar juicer_tools.jar
至此,juicer的安装就基本完成了
-
juicer的使用
运行下面的代码,记得要根据自己的实际情况修改路径。
~/nw/juicer/scripts/juicer.sh \
-z ~/nw/juicer/references/Homo_sapiens_assembly19.fasta#参考序列 \
-p ~//nw/juicer/restriction_sites/hg19.chrom.sizes#染色体size \
-y ~/nw/juicer/restriction_sites/hg19_MboI.txt#限制位点 \
-d ~/nw/rawdata/A549/replicate1 #hi-c数据\
-D ~/nw/juicer#工作目录
Usage: juicer.sh [-g genomeID] [-d topDir] [-q queue] [-l long queue] [-s site]
[-a about] [-S stage] [-p chrom.sizes path]
[-y restriction site file] [-z reference genome file]
[-C chunk size] [-D Juicer scripts directory]
[-Q queue time limit] [-L long queue time limit] [-b ligation] [-t threads]
[-A account name] [-e] [-h] [-f] [-j]
#-g 指定参考序列,默认是hg19
#-s 限制性内切酶("HindIII" or "MboI")
#-t 运行BWA时的线程数
-
结果输出
结果文件都放在了生成的 aligned 文件夹中,主要文件是inter.hic和inter_30.hic文件,其中的inter_30.hic 是设置了 MAPQ threshold >30 后得到的结果。
-
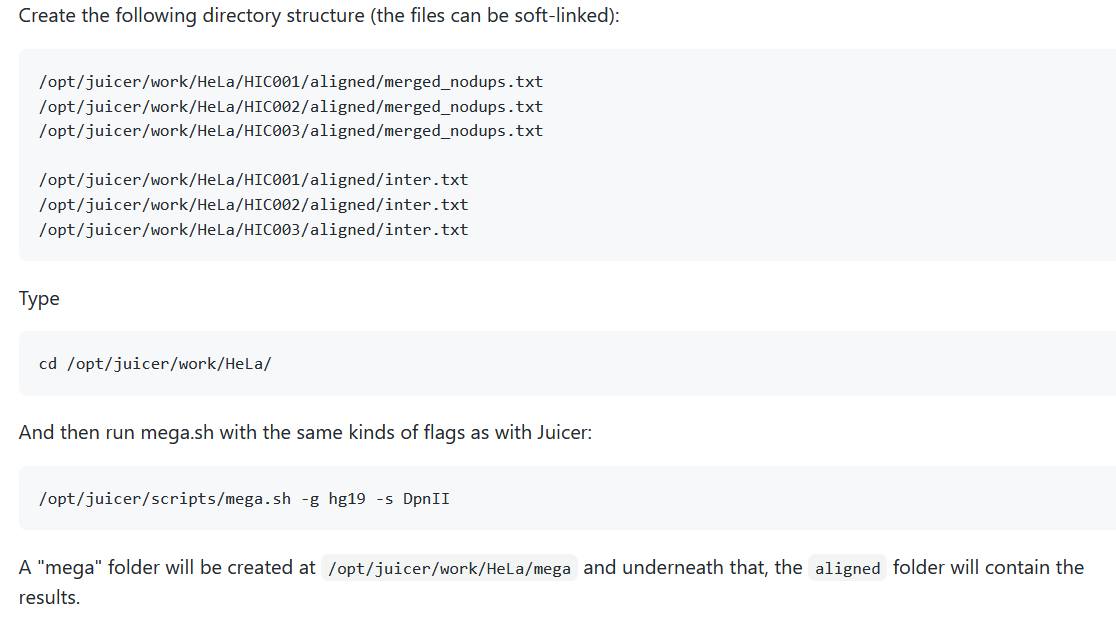
重复样本合并
-
使用juicer_tools工具进行下游分析
目前针对Hi-C数据的研究主要是三个方面,分别是A/B comparment ,TADS,Loops。
juicer_tools.jar 功能介绍
arrowhead 注释TAD
hiccups 注释loop
motigs 定位CTCF元件
hiccupsdiff 从多个loos文件中找到不同的loop
apa 聚合峰的分析
pearsons 计算O/E的皮尔森相关系数
eigenvector 计算特征向量的皮尔森相关系数
dump .hic文件互作矩阵提取
pre 非juicer数据转.hic文件
-
Arrowhead
arrowhead [-c chromosome(s)] [-m matrix size] [-r resolution]
[-k normalization (NONE/VC/VC_SQRT/KR)] <HiC file>
<output_file> [feature_list] [control_list]
| 参数 | 描述 |
|---|---|
| 必须参数 | — |
| Juicer生成的.hic文件 | |
| contact domain文件, 可导入Juicebox 通过2D annotation进行可视化 | |
| 可选参数 | — |
| -c | 染色体, 多个染色体写法chr1,chr2,chr3, 或者是1,2,3 |
| -m | 沿着对角线移动的窗口, 必须为偶数,移动步距是m/2, 默认为2000 |
| -r | 查找的分辨率, 默认为10k, 设置值取决于.hic文件 |
| -k | 选择标准化的方式, <NONE/VC/VC_SQRT/KR>, 通常选KR |
- 输出格式
chromosome1 x1 x2 chromosome2 y1 y2 color corner_score Uvar Lvar Usign Lsign
chromosome 染色体
x1 = y1, x2 = y2 区域范围
corner_score, contact domain 是边缘bin的可能性, 值越大, 可能性越高
Uvar, 上三角形的方差
Lvar, 下三角形的方差
Usign, 上三角形sign of the entries的总和
Lsign, 下三角形sign of the entries的总和
-
hiccups
识别染色质环的HICCUPs算法必须通过GPU加速运行才可以,所以没有安装GPU卡的普通服务器无法运行这个步骤。但我们可以使用cpu版本的HICCUPS算法,与GPU相比,识别的loop数目变少了。
hiccups [-m matrixSize] [-c chromosome(s)] [-r resolution(s)]
[-k normalization (NONE/VC/VC_SQRT/KR)] [-f fdr]
[-p peak width] [-i window] [-t thresholds]
[-d centroid distances] <HiC file> <outputDirectory> [specified_loop_list]
| 参数 | 描述 |
|---|---|
| 必须参数 | — |
| –cpu | CPU版本必选 |
| MAPQ>30的.hic文件 | |
| 计算的结果文件和中间文件 | |
| 可选参数 | — |
| –restrict | 使用GPU版本跑CPU版本的参数信息, 用于比较两者的差异 |
| [specified_loop_list] | 位置的可选参数, 对应于merge的loop文件, 用于返回特定loop |
| -m | 我理解为GPU并行的线程数, 不会影响结果, 数值越大, 速度越快, 独显可设置500, 100 或2048, 集成选卡最好不要超过100 |
| -c | 染色体, 多个染色体写法chr1,chr2,chr3或者1,2,3 |
| -r | 分辨率, 多个分辨率用’,’隔开, 不同的分辨率再设置其他参数时也需用’,’隔开 |
| -k | 选择标准化的方式, <NONE/VC/VC_SQRT/KR>, 通常选KR |
| -f | FDRvalues |
| -p | 峰的宽度 |
| -i | 窗口的宽度 |
| -d | Distances used for merging nearby pixels to a centroid |
| -t | 四个用逗号隔开的参数, 用于不同分辨率的loop合并的门槛 |
-
Eigenvector
eigenvector可用于在高分辨率的Hi-C数据中描绘区室;特征向量的符号通常表示区室。特征向量是皮尔逊矩阵的第一主成分
java -jar juicer_tools.jar eigenvector KR HIC001.hic 1 BP 1000000
eigenvector <NONE/VC/VC_SQRT/KR> <hicFile(s)> <chr> <BP/FRAG> <binsize> [outfile]
6,Hi-C数据可视化
-
juicerbox可视化,既可以在本地使用,也可以在线使用。这也是juicer相比其他软件的优点。我就介绍下线上版本的使用:
- https://aidenlab.org/juicebox/
-
加载.hic文件
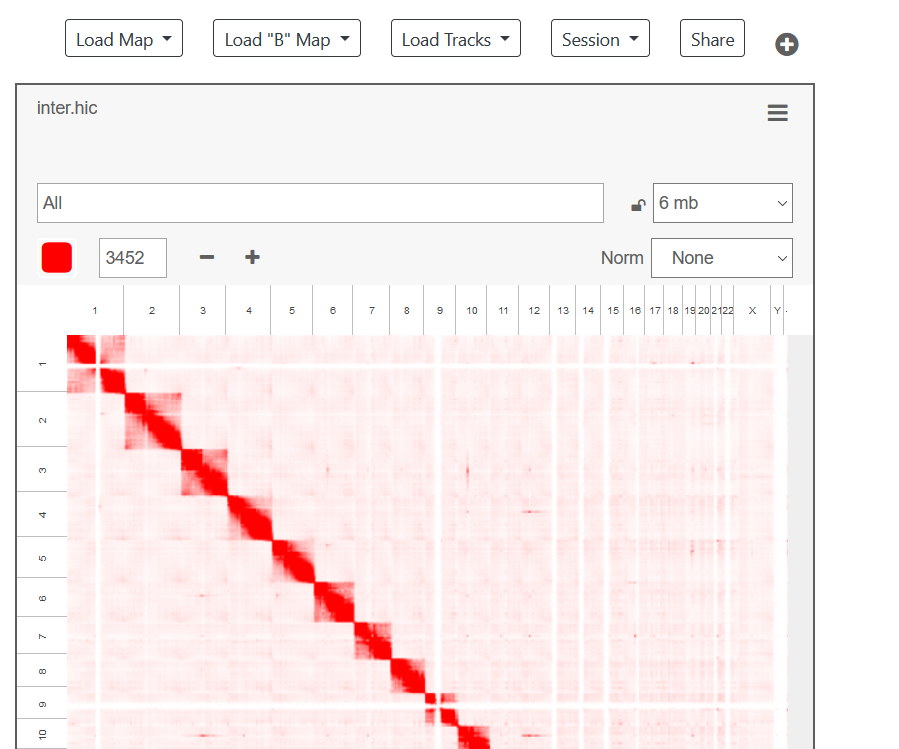
-
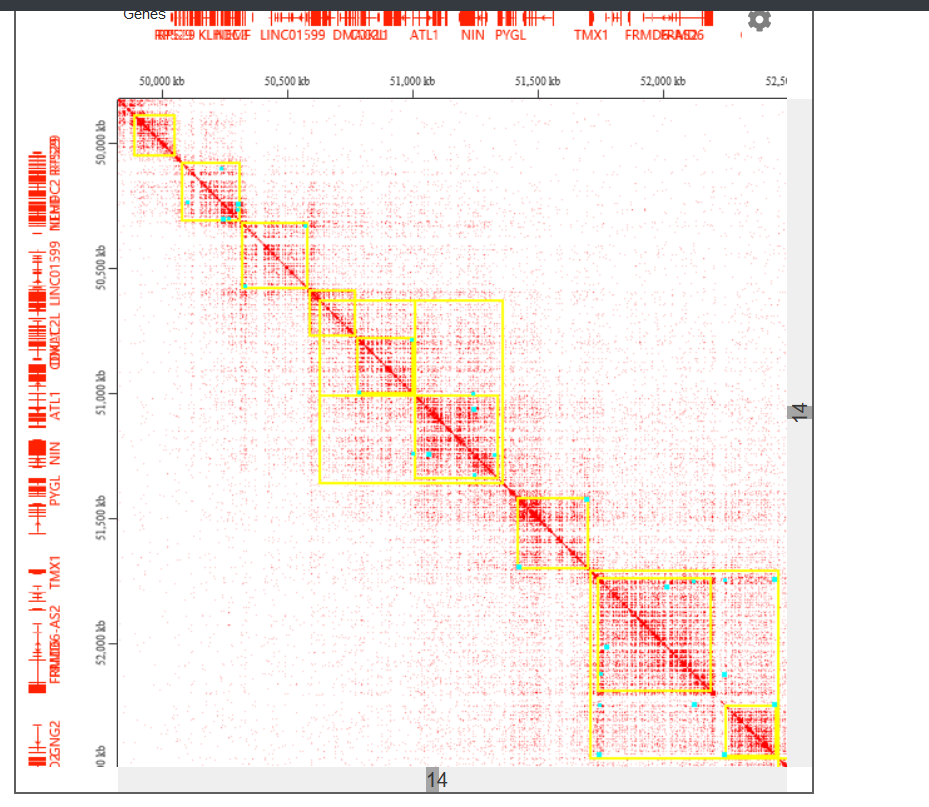
加载一维注释
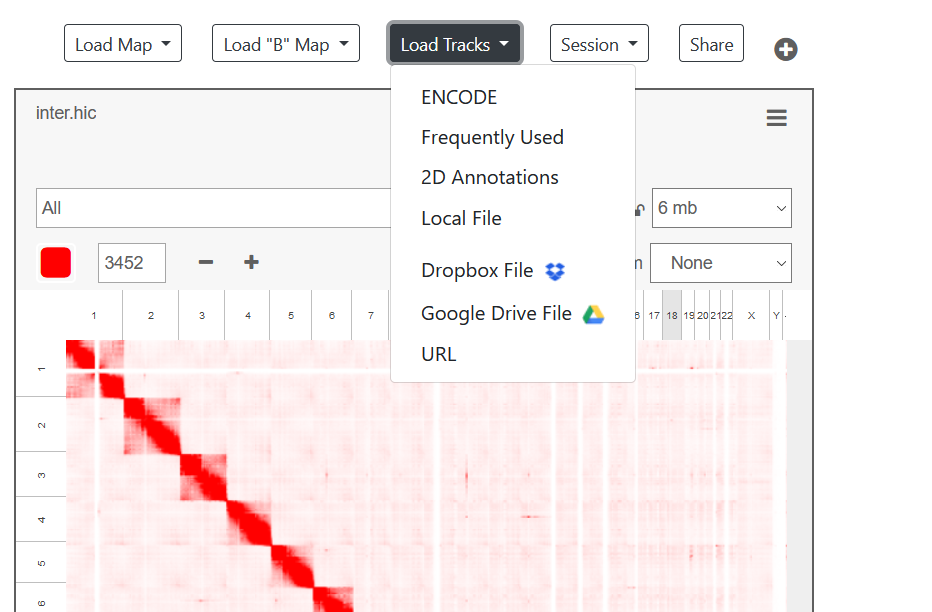
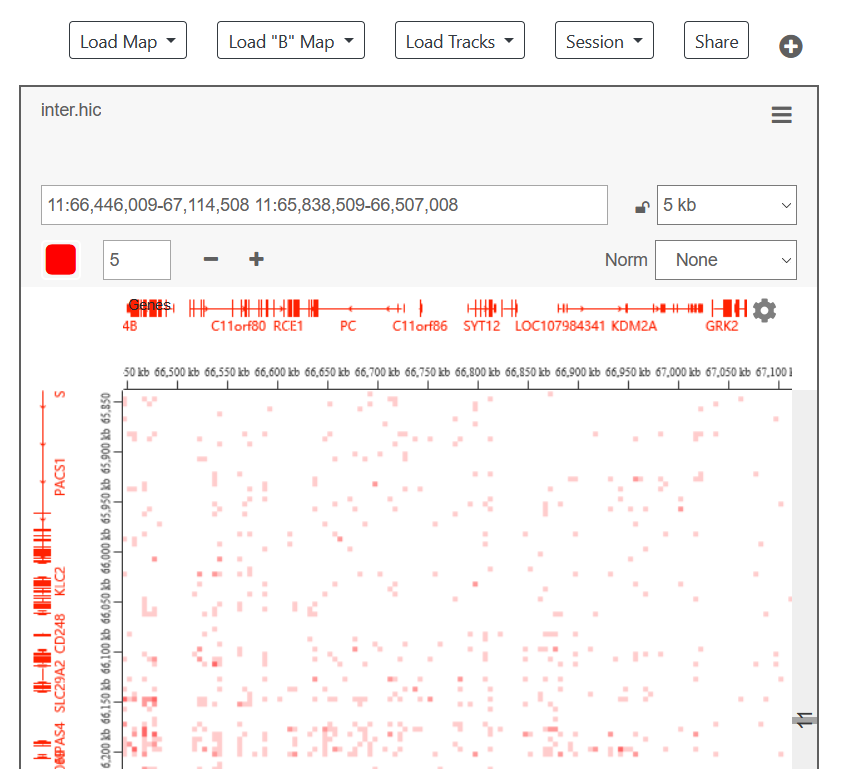
-
加载二维注释,黄的的是TAD,天蓝色的是loop